SEquence CApture PRocessor (SECAPR)
![]()

Original Publication: https://doi.org/10.7717/peerj.5175
We are now teaching a 1-week intensive course on target enrichment data, including practical exercises for all functionalities of the SECAPR pipeline. Check the ForBio webpage for the next open course dates and for information about past courses.
Real data tutorial (incl. installation) click here
Detailed documentation of all functions click here
Overview
This semi-automated pipeline aims to make the processing and analysis of sequence capture (= target enrichment) data simple and straight forward for all users. The detailed documentation and simple installation makes this pipeline accessible also for users with limited biofinformatic knowledge, while enabling user-defined processing options for the more experienced users.
We included an empirical data tutorial in the pipeline documentation, which covers the processing from raw Illumina read data into multiple seqeunce alignments (MSAs) for phylogenetic analyses, including the compiling of allele sequence MSAs. This workflow can be applied to any Illumina dataset, independently of the underlying bait set and organism group.
Some functions in this pipeline are inspired by scripts from the Phyluce pipeline by Braint Faircloth, which is aimed at the processing of Ultraconserved Elements (UCEs). To honour some of the ideas belonging to Brant Faircloth, and the generous sharing of all his code as open-source, we ask you to cite the Phyluce pipeline (Faircloth 2016) alongside with ours (Andermann et al. 2018), when using SECAPR.
Workflow
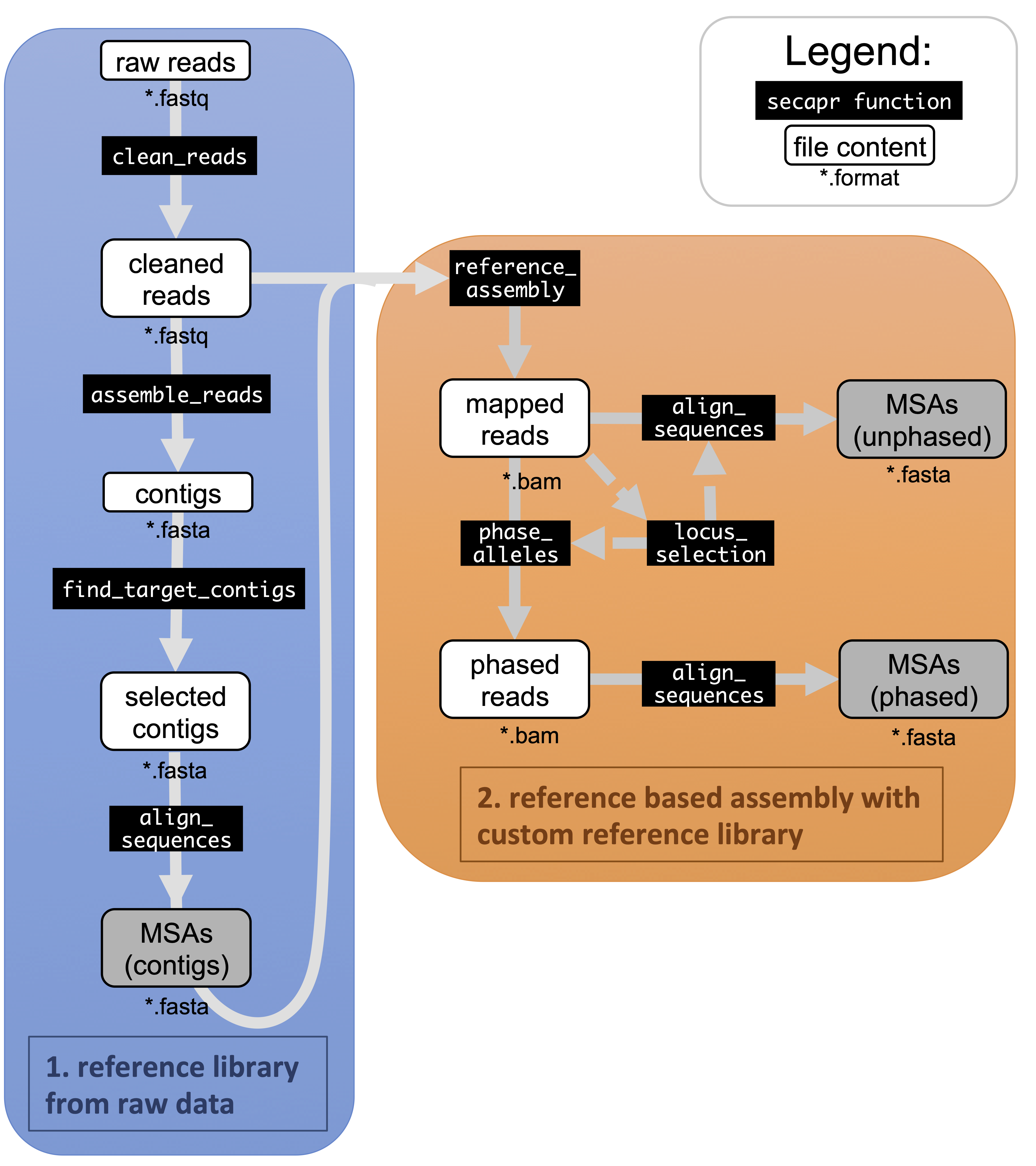
SECAPR analytical workflow. The flowchart shows the basic SECAPR functions, which are separated into two separate steps (colored boxes). Blue box (1. reference library from raw data): in this step the raw reads are cleaned and assembled into contigs (de novo assembly); Orange box (2. reference based assembly with custom reference library): the contigs from the previous step are used for reference-based assembly, enabling allele phasing and additional quality control options, e.g. concerning read-coverage. Black boxes show SECAPR commands and white boxes represent the input and output data of the respective function. Boxes marked in grey represent multiple sequence alignments (MSAs) generated with SECAPR, which can be used for phylogenetic inference.
Please cite:
Andermann T., Cano Á., Zizka A., Bacon C., Antonelli A. 2018. SECAPR—a bioinformatics pipeline for the rapid and user-friendly processing of targeted enriched Illumina sequences, from raw reads to alignments. PeerJ 6:e5175 https://doi.org/10.7717/peerj.5175
Faircloth B.C., PHYLUCE is a software package for the analysis of conserved genomic loci, Bioinformatics, Volume 32, Issue 5, 1 March 2016, Pages 786–788, https://doi.org/10.1093/bioinformatics/btv646